Protein structure basically depends on the arrangement of different amino acid residues in a three-dimensional way.
In general, proteins are macromolecules and have four different levels of structure.
What are the four levels of protein structure?
1) Primary structure: It represents the linear amino acid sequence.
2) Secondary structure: local folding of amino acids, mainly in the primary structure.
3) Tertiary structure: global folding of a single peptide strand.
4) Quaternary structure: Interactions between two individual subunits lead to covalent bonding of the two subunits, and that generates functional proteins.
The primary structure of the protein
- Protein primary structure represents the linear sequence (order) of the amino acid units in an entire protein molecule.
- The primary structure is stabilized only by covalent peptide bonds, which offer metastability to proteins and show a higher level of stability even when heated at 1800C.
- This is the first structural level of proteins during protein structure formation, and it also shows the final structural level of proteins when a protein is subjected to complete denaturation.
- It means a protein consisting of any type of structure (Secondary, Tertiary, or Quaternary), finally reaches the primary structural level if you do complete denaturation.
Read Anfinsen experiment
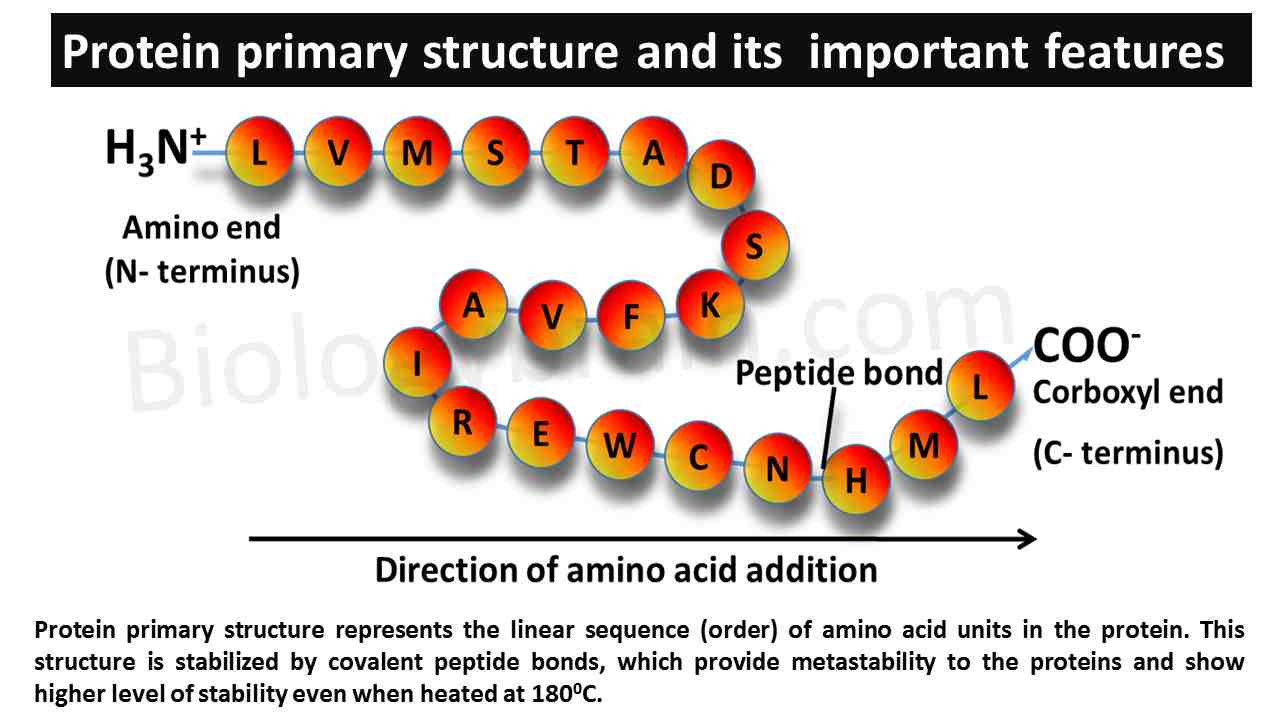
- The primary structure shows a distinct feature, such as polarity because one end of the protein contains a free α-amino group and another end of the protein contains a free carboxylic group.
- Based on this polarity, the ends of the primary structure are represented as N- terminus (α-amino group side) and C- terminus (carboxylic end).
- This structure determines all other structural levels of proteins. The amino acid sequence of this structure also determines the codon sequence in the coding gene of a particular protein.
- Any alterations in the codon sequence of the gene can affect the primary structure of a protein by changing the amino acid sequence.
- These changes contribute to functional changes in the protein, that result in the development of harmful effects on the body.
Secondary structure of the protein
- In the process of protein synthesis, the term “secondary structure” corresponds to the local folding of the protein at the primary structure level.
- In the primary structure, the closely arranged regions will come together to form weak interactions by hydrogen bonding and generate the secondary structure of a protein.
- It is the only structural level in which covalent interactions are not present, but hydrogen bonds can form the noncovalent interactions in the local folding.
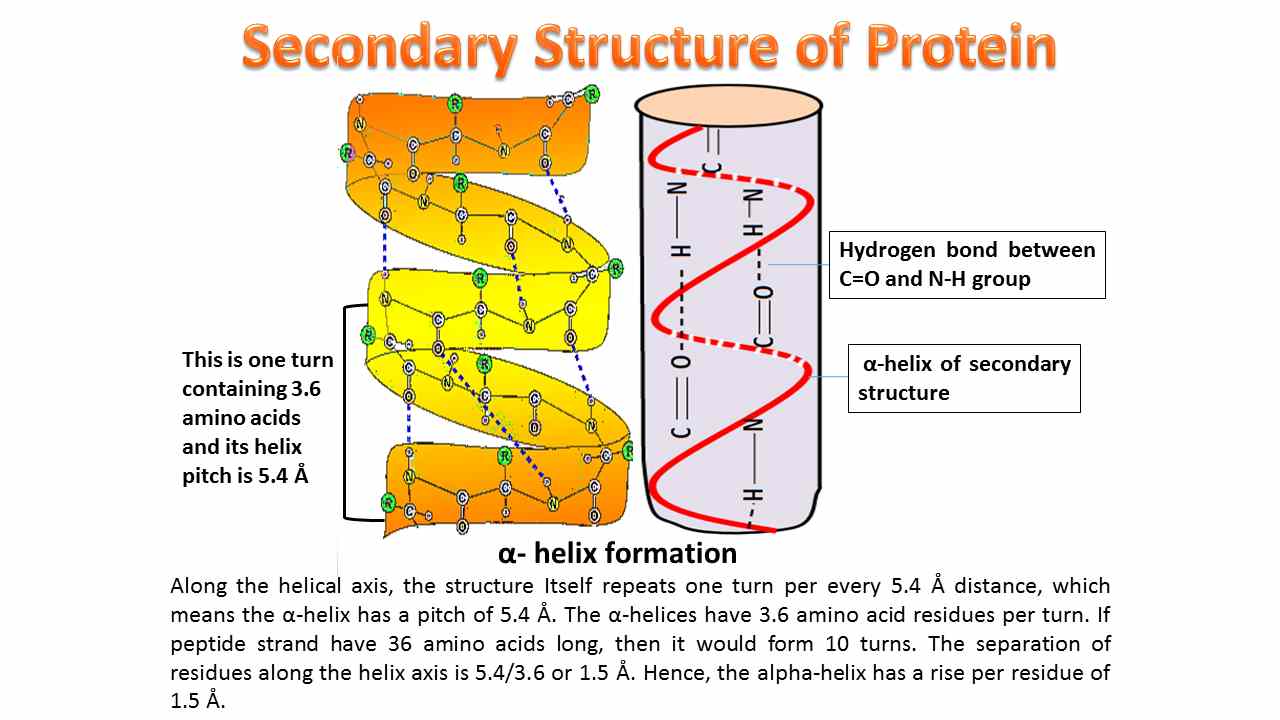
- The secondary structure of a protein is very important to fulfill the functional characteristics of the protein.
- The secondary structure of the protein is solely stabilized by hydrogen bonds.
- The hydrogen bonding will take place mainly between the carboxyl oxygen of one peptide bond and the imino hydrogen of another peptide bond.
- The main purpose of the formation of secondary structures is to reduce the polarity associated with peptide bonds so that higher-order structures can be generated.
- CD spectroscopy can be used to study the secondary structure of the proteins and the Ramachandran plot can be used to determine the feasibility of the occurrence of secondary structure.
- α-helix was the first-ever secondary structure proposed by Linus Pauling in 1951. The β- pleated sheet was the regular secondary structure proposed by Linus Pauling and Robert Corey in 1952.
- Both the structures, α-helix, and β- pleated sheets were published in “The General of Biological Chemistry”.
- These two forms show regular secondary structural levels, which are well-known for many functional proteins.
- The common irregular secondary structure is the random coil form.
Helical formation of protein
The amino acids in the protein structure will be arranged in linear passion. However, in biological conditions, sequences of amino acids will fold regularly or irregularly by molecular attractions to give suitable structure to the protein to become metabolically active.
Types of regular right-handed α-helix
Basically, three types of regular right-handed helical structures have been studied, such as α-helix, 310 helix, and π (pi) helix.
Alpha helix form
- Involves intrachain hydrogen bonding and the most common α-helix is right-handed and contains 3.6 amino acid residues per turn. The distance between adjacent amino acid residues is known as helical rise and the distance will be 1.5Å or 15nm and helical pitch of 5.4 Å (0.54nm).
Helical pitch = (Helical rise per residue × No of residues in a loop).
- For every amino acid residue, the helix turns by 1000. The helical repeat occurs every 5 turns or 18 amino acids, which means the same kind of amino acids is repeated for every 5 turns in a single peptide strand.
- In the regular right-handed α-helix, we can find a hydrogen-bonded loop, which will be formed by hydrogen bonding between carboxyl oxygen of Ath amino acid and imino hydrogen of A+ 4th amino acid.
- Every hydrogen-bonded loop contains 13 atoms, therefore this regular right-handed α-helix is represented as 3.613.
- The aromatic amino acid, proline strongly de-stabilizes the regular α-helix because the imino nitrogen of proline lacks hydrogen atom for the formation of the hydrogen bond. Hence, the un-bonding nature of the proline disturbs the α-helix formation. If proline is incorporated into the peptide sequence, then it forms a kink in the protein. In a few cases, by chance, if proline is present, then it completely collapses the functional properties of the proteins.
- Glycine is the simple and basic structure of amino acid, which strongly de-stabilizes the α-helix, because it contains only hydrogen atoms as a highly flexible side chain, hence it causes immediate disturbance in the regular turns of the polypeptide. Therefore, this amino acid is represented as a hinge amino acid.
- While, the amino acids such as arginine and aspartate contain high polarity in their side chains; hence, they also disturb the regular α-helix.
- However, hydrophobic amino acids such as alanine, valine, leucine, methionine, etc. are the most suitable amino acids for the formation of regular α-helix.
- The best examples for α-helix contained protein structures include α-keratin, myoglobin, and hemoglobin.
310-helix
- This helix contains three amino acids per turn and ten atoms per hydrogen-bonded loop.
- In 310 helices, the hydrogen bond can be formed between the carbonyl oxygen of ith of residue and imino hydrogen of i+3rd amino acid residue (H bond pattern between i and i+3).
- This loop contains 3 residues per turn, consecutive amino acids make an angle of 120° around the helical axis, a helical rise per amino acid of 2 Å, and a helical pitch of 5.8-6 Å (helical rise per residue × No of residues in a loop).
pi helix
- pi helix (π–helix) is another type of secondary structure present in some proteins. 15% of known protein structures contain these short pi–helices. Biologists believe that the formation of the pi helix is due to evolutionary adaptations derived from a single amino acid insertion into an α-helix.
- π helix or 4.416 helix contains 4.4 amino acids per turn and 16 atoms in the hydrogen-bonded loop.
- The hydrogen-bonded loop can be formed by hydrogen bonding between carboxyl oxygen of Ath amino acid and imino hydrogen of A+5th amino acid.
Beta pleated sheet
- The hydrogen bond formation in the β-pleated sheet is opposite to the α-helix. In an α-helix, the hydrogen bond formation will be within the same chain (intrachain bonding), but in a β-pleated sheet, hydrogen bond formation will be between two chains (interchain bonding).
- However, in some cases, bonding can be intrachain, which forms an antiparallel β-pleated sheet when a respective chain folds back itself to form a hydrogen bond.
- The two chains involved in β-pleated sheet formation are called β-strands. Based on the two β-strand arrangements, the β-pleated sheet has been divided into two forms, parallel and antiparallel.
Parallel beta-pleated sheet
- The hydrogen bond formation in parallel β-pleated sheet strictly between interchains.
- In a parallel β-pleated sheet, the two β-strands are arranged in the same directions. It means the amino terminus of one strand will be closed to the amino terminus of the opposite strand.
- Similarly, the C- terminus of one strand will be closed to the C-terminus of another strand. However, this closer proximity of similar groups in the strands develops an intrinsic repulsion, which leads to a reduction in structural stability.
- The hydrogen bonds between the chains are not compact because an amino acid in one chain forms hydrogen bonds with two different amino acids in the second chain.
Antiparallel beta-pleated sheet
- The hydrogen bond formation in an antiparallel β-pleated sheet can be formed between interchains or within intrachain.
- In an antiparallel β-pleated sheet, the two β-strands are arranged in opposite directions. The bond formation occurs between the amino groups of one strand and the closely located carboxyl terminus of the other strands.
- The hydrogen bonds are highly compact because amino acid in one chain forms the hydrogen bonds with single amino acid in the second chain.
- The commercially valuable insoluble proteins of silk, such as fibroin, sericin, etc. are the best examples of the antiparallel β-pleated sheet-contained proteins.
- Interestingly, in addition to parallel and anti antiparallel β-sheets, the mixed β-sheets are also possible. It means, we can see both types of above-mentioned sheets in mixed β-sheets. Mixed β-sheets are only possible when a β-sheet or protein contains three or more β-strands.
The tertiary structure of the protein
- The tertiary structure corresponds to the protein folding or three-dimensional (3D) structure.
- Tertiary structure is the next level of the structure after secondary structure formation.
- In this structure, the functional region of the protein will be generated when faraway, separated amino acid residues in the peptide come together and form a bond.
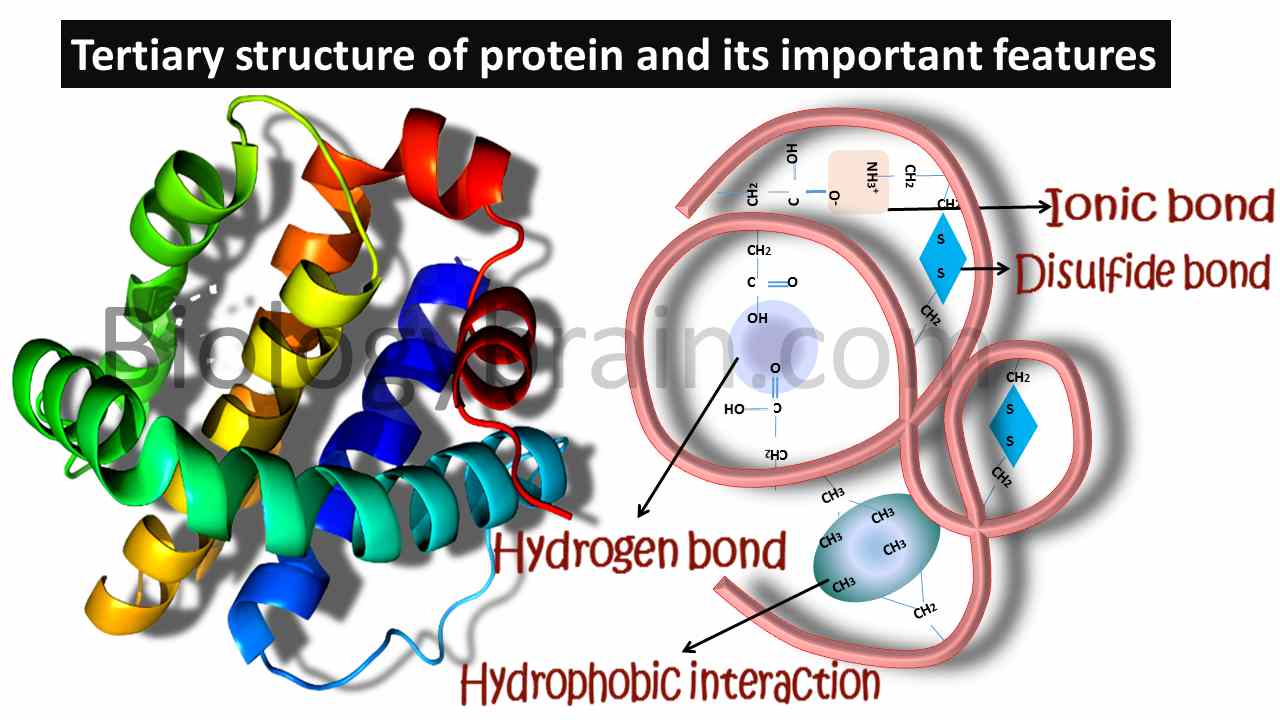
- A protein having a properly formed 3D structure is called a native protein. This native structure of the protein will have complete catalytic activity.
- In any circumstances, if protein losses the 3D structure then that protein is represented as denatured protein. This denatured protein completely losses the catalytic activity.
- The tertiary structure of the protein will be formed by both covalent and noncovalent interactions. The only possible covalent interactions can be noticed in disulfide bridges, which are formed between sulfur-containing amino acids.
- Noncovalent interactions can be seen in both salt bridges (ionic bonds) and hydrophobic interactions.
- Salt bridges will be formed between negatively charged and positively charged side chain-containing amino acids, whereas hydrophobic interactions will be formed between hydrophobic amino acids.
- Secretary and extracellular proteins contain both covalent and noncovalent interactions, but intracellular proteins are stabilized by purely noncovalent interactions since the cytoplasm is a reducing environment, where the covalent interactions are unstable.
- The covalent interactions of protein can be seen in the form of intra-subunit or inter-chain disulfide bridges. The amino acid cysteine, found in the same protein molecule is the only contributor to forming the disulfide bridge by the oxidation process.
- The higher-order tertiary structure of proteins shows more stability due to the presence of disulfide bridges which are bound by covalent interactions.
- Disulfide bridges are responsible for greater stability of extracellular proteins compare to intracellular proteins, because intracellular proteins present in a highly reducing environment (cytoplasm).
- In the cytoplasm, since the formation of strong disulfide brides are not possible, then the only noncovalent interactions stabilize the structure of intracellular proteins.
- Even though so many noncovalent interactions are present in the protein structure, only hydrophobic noncovalent interactions provide the driving force for protein folding.
- The tendency of hydrophobic side chains of the amino acids to repel from the water molecule leads to the gathering of all side chains in one place and forming hydrophobic interactions. However, these are only interactions, but not true bonds.
- The gathering of all hydrophobic side chains at one region in a protein is called the nucleation center, these nucleation centers can be seen in different regions of the protein.
- A nucleation center is a region in a protein where folding is initiated. Often, such nucleation centers constitute domains. A domain is any region in a protein structure that is capable of independent folding. Interestingly, domains are generated only in tertiary structures.
- The sulfur group-containing amino acid methionine, aromatic amino acids phenylalanine and tryptophan, and branched-chain amino acids, valine, isoleucine, and leucine are considered hydrophobic amino acids and form hydrophobic interactions.
- The salt bridges are another kind of noncovalent interaction.
- The salt bridges will form between the positively charged side chain of lysine or arginine and the negatively charged side chain of aspartate or glutamate.
- Hydrogen bonds are the third kind of noncovalent bonds formed by side chains of imino group-containing amino acids, asparagine and glutamine, and hydroxyl group-containing amino acids serine and threonine.
- The hydrogen bonds and salt bridges can be easily disrupted compared to hydrophobic interactions, therefore the conformational changes in a protein can be involved in the temporary loss of salt bridges and hydrogen bonds.
The quaternary structure of the protein
- Most protein structures are made up of a single polypeptide chain. However, some proteins are formed by more than one polypeptide chain (subunits) by interchain interactions known as quaternary structures. Subunits of the quaternary structure are sometimes identical, and a few times they are unique to each other.
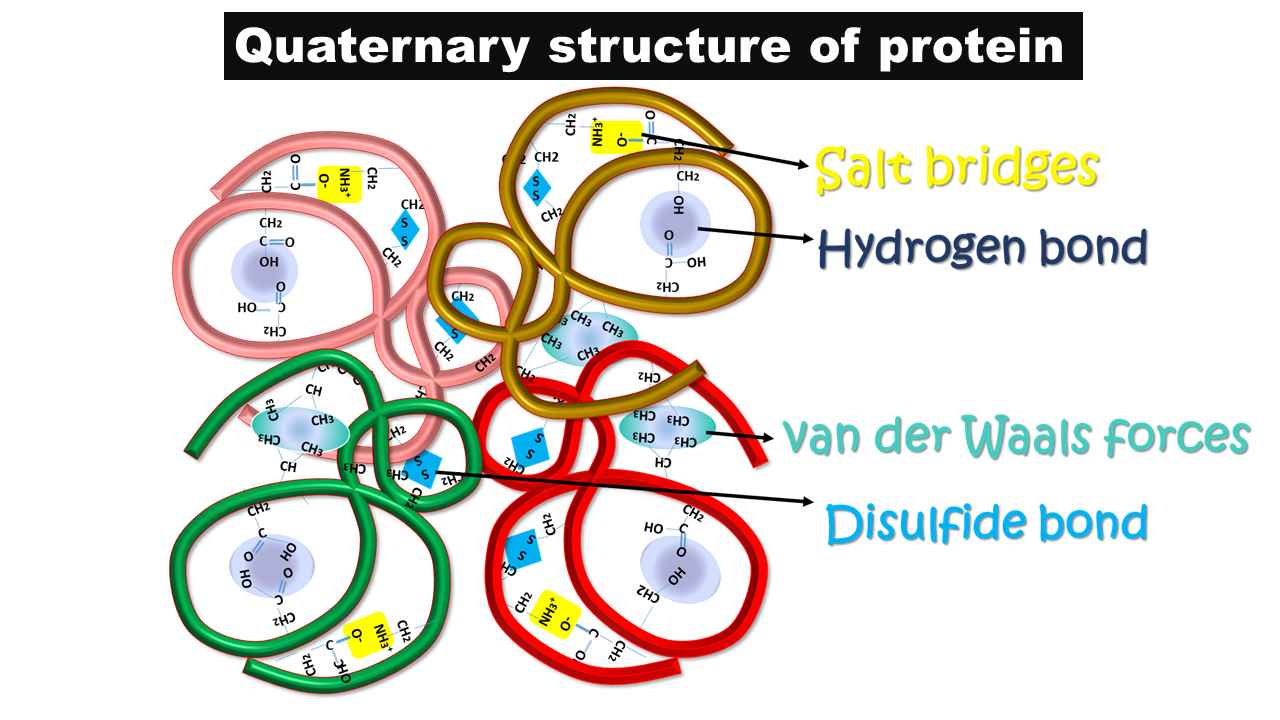
- For example, the dimer is the simplest form of quaternary structure, that contains two identical subunits. This type of organization can be observed in the Cro protein (DNA-binding protein) of bacteriophage λ. And, the hemoglobin is a tetrameric form, which is another example of a tertiary structure, that contains two distinct types of peptides. Two subunits of one type (designated α) and two subunits of another type (designated β).
- The interactions between multi-polypeptide chains form a quaternary structure more stable.
- The quaternary structure of the protein is possible only for multisubunit proteins, which contain intersubunit interactions.
- Whatever forces stabilizing the subunits of the quaternary structure are similar to the forces stabilizing the tertiary structure.
- The quaternary structure of the protein is stabilized by both covalent and noncovalent interactions.
- As noncovalent interactions, the interchain salt bridges are present.
- These interactions are formed between side chains of positively and negatively charged amino acids.
- Example: The extracellular protein collagen is crucial for maintaining the structural integrity of many tissues in the body, such as blood vessels, tendons, bones, and ligaments. The salt bridges between basic amino acid, lysine, and acidic amino acid, glutamate in the peptide chains of collagen protein stabilize its structure.
- As a covalent interaction, the thiol group-containing amino acid, cysteine residue from two different peptide chains synthesizes disulfide bonds between these two inter subunits and generates more stability for the structure.
- These disulfide bonds are the only covalent interactions possible in the quaternary structure.
- In a few cases, hydrophobic interactions and hydrogen bonds are also possible as noncovalent interactions and contribute to the structural stability of the protein.
An example of quaternary structure:
- Insulin is an extracellular protein that has been stabilized by both covalent and noncovalent interactions.
- Whereas intracellular proteins, such as hemoglobin, can only have noncovalent interactions.
- In the case of insulin, two interchain disulfide bridges are found between chains a and b.
- In the case of hemoglobin, the quaternary structure began with the formation of a heterodimer of α and β subunits, which will be held together strongly by hydrophobic interactions and will remain intact even when conformational changes occur in hemoglobin.
- Two such heterodimers come together to form the heterotetramer, in which similar kinds of subunits interact through weak salt bridges that can be disrupted when hemoglobin undergoes conformational changes due to ligand binding.
- Considering the similarity in each subunit, hemoglobin was considered myoglobin in muscle cells, and the structure of hemoglobin was represented as mb-mb2.
Read more:
How do enzymes speed up chemical reactions?
Lysosomal enzymes list and their function
What is a GPCR (G-protein coupled receptor)? Definition and Types
Thermodynamics of Protein Folding
Thermogenin, an uncoupling protein-Definition, and Function
The characteristics of enzymes
References:
- The Shape and Structure of Proteins. Molecular Biology of the Cell. 4th edition.(%https://www.ncbi.nlm.nih.gov/books/NBK26830/).
- Sun PD, Foster CE, Boyington JC. Overview of protein structural and functional folds. Curr Protoc Protein Sci. 2004 May;Chapter 17(1):Unit 17.1. doi: 10.1002/0471140864.ps1701s35.
- Chapter 3Protein Structure and Function. Biochemistry. 5th edition (%https://www.ncbi.nlm.nih.gov/books/NBK21177/).
- Chapter A06, Protein Structure, Modelling and Applications. Bioinformatics in Tropical Disease Research: A Practical and Case-Study Approach (%https://www.ncbi.nlm.nih.gov/books/NBK6824/).
- Biochemistry, Primary Protein Structure. Treasure Island (FL): StatPearls Publishing; 2021 Jan (%https://www.ncbi.nlm.nih.gov/books/NBK564343/).
- Marks DS, Hopf TA, Sander C. Protein structure prediction from sequence variation. Nat Biotechnol. 2012 Nov;30(11):1072-80. doi: 10.1038/nbt.2419.
- Kuhlman B, Bradley P. Advances in protein structure prediction and design. Nat Rev Mol Cell Biol. 2019 Nov;20(11):681-697. doi: 10.1038/s41580-019-0163-x. Epub 2019 Aug 15.
- Section 3.5Quaternary Structure: Polypeptide Chains Can Assemble Into Multisubunit Structures. Biochemistry. 5th edition.